
Start by selecting the type of study that you plan to submit:
- Single Site Study
- Multi-Site Study (Pitt as IRB of Record)
- Multi-Site Study (Pitt cedes IRB review to another IRB)
Single Site Study
- Study has one participating site
- Pitt is the IRB of Record
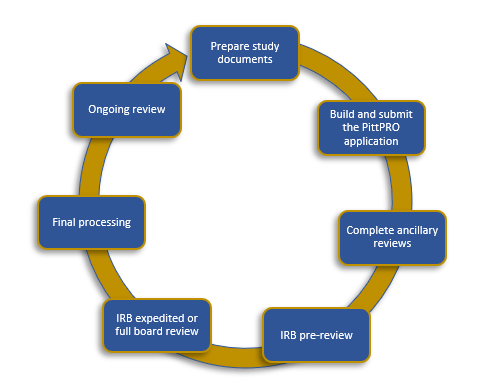
Submission process steps:
1. Prepare study documents: ensure your application is complete and through
2. Build and submit the PittPRO application: contact askirb@pitt.edu to schedule a PittPRO training course if you’re unfamiliar
3. Ancillary Reviews: Review Chapter 8 of the IRB P&P for more information
4. IRB Pre-review: This is the first IRB review. An IRB staff member will review the application for common errors and request clarifications/revisions as needed
5.IRB Review:
- Full Board: protocol appears before a convened IRB and is reviewed by members who consider the approval status
- Administrative Review: protocol is reviewed by a member of the Exempt/Expedited Team who will correspond directly with you to secure approval
6. Final processing: IRB staff is completing final regulatory documentation, and finalizing documents, including the approval letter
7. Ongoing review:
- Modifications to an approved study must be submitted to the IRB prior to implementation
- Continuing Review (i.e. renewals): Greater than minimal risk and some minimal risk protocols must be reviewed at least annually (prior to the named expiration date), per the Federal regulations. Contact askirb@pitt.edu if you are unsure if your protocol requires continuing review
Multi-Site Research where Pitt is IRB of Record
- Multiple participating sites
- Pitt IRB will act as IRB of Record for all sites
- Contact the Reliance Team at irb.reliance@pitt.edu to discuss the submission
Submission Process Steps:
1. Complete an entry in the Reliance Request System: This is the first step in research involving reliance or a Single IRB (sIRB) mechanism for multi-site research. Learn more about the questions in the Reliance Request System
2. Prepare study documents: ensure your application is complete and through
3. Build and submit the PittPRO application: contact askirb@pitt.edu to schedule a PittPRO training course if you’re unfamiliar
4. Ancillary Reviews: Review Chapter 8 of the IRB P&P for more information
5. IRB Pre-review: first IRB review. An IRB staff member will review the application for common errors and request clarifications/revisions as needed
6. IRB Review:
- Full Board: protocol appears before a convened IRB and is reviewed by members who consider the approval status
- Administrative Review: protocol is reviewed by a member of the Exempt/Expedited Team who will correspond directly with you to secure approval
7. Final processing: IRB staff is completing final regulatory documentation, and finalizing documents, including the approval letter
8. Ongoing review:
- Modifications to an approved study must be submitted to the IRB prior to implementation
- Continuing Review (i.e. renewals): Greater than minimal risk and some minimal risk protocols must be reviewed at least annually (prior to the named expiration date), per the Federal regulations. Contact askirb@pitt.edu if you are unsure if your protocol requires continuing review
Multi-Site Research where Pitt cedes IRB Review
- Multiple participating sites
- Pitt IRB will cedes IRB review authority to another IRB
- Contact the Reliance Team at irb.reliance@pitt.edu to discuss the submission
Submission Process Steps
1. Complete an entry in the Reliance Request System: This is the first step in research involving reliance or a Single IRB (sIRB) mechanism for multi-site research. Learn more about the questions in the Reliance Request System
2. Prepare study documents: ensure your application is complete and through
3. Build and submit the PittPRO application: contact askirb@pitt.edu to schedule a PittPRO training course if you’re unfamiliar
4. Ancillary Reviews: Review Chapter 8 of the IRB P&P for more information
5. IRB HRPP Review: the Reliance Team will complete the required local reviews
6. Final processing: IRB staff is completing final regulatory documentation, and finalizing documents, including the approval letter
7. Ongoing review:
- Modifications to an approved study must be submitted to the IRB prior to implementation
- Continuing Review (i.e. renewals): Greater than minimal risk and some minimal risk protocols must be reviewed at least annually (prior to the named expiration date), per the Federal regulations. Contact askirb@pitt.edu if you are unsure if your protocol requires continuing review
Thank you to the University of Utah Institutional Review Board, which has graciously provided this content.